Projects in the Schwarz Lab
Current Projects in the lab can be grouped into three categories:
1. Synapse Formation at the Drosophila Neuromuscular Junction,2. The Role of Ral and the Exocyst in Synaptic Plasticity, and
3. The Transport of Mitochondria in Axons and Dendrites and their Involvement in Neurodegenerative Disease.
Synapse Formation at the Drosophila Neuromuscular Junction:
The fly neuromuscular junction has long served as a model for synapse development and function. Each axon that innervates the muscles of the larval body wall acquires a characteristic anatomical shape. With boutons that release glutamate and give rise to excitatory synaptic potentials that sum in a graded fashion, it has many features that resemble the excitatory synapses of the mammalian CNS. There are also more than 3 decades of electrophysiological studies describing the functional properties of the synapse, and a wealth of genetic studies that have elucidated pathways regulating the size and strength of the synapse. It also serves as a model of synaptic growth and plasticity because of the impressive increase in bouton number that occurs between the time when the first-instar larva emerges from the egg case and its ultimate form in late third-instar larvae.
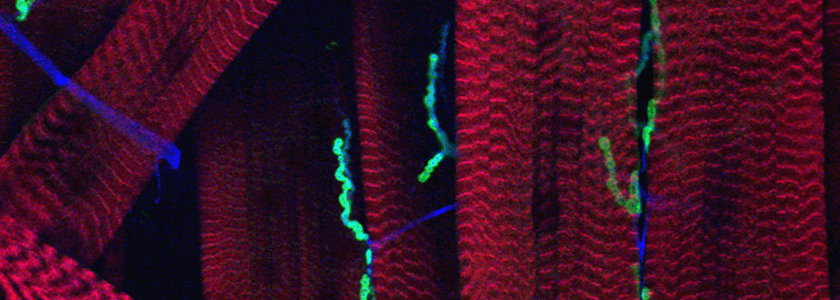
Our current projects arose from a mutant screen for defects in synaptic transmission carried out by Dion Dickman when he was a student in the lab. One set of mutations fell in the gene for an importin family member. The finding initiated a series of studies concerned with importin function presynaptically and postsynaptically, including how signals generated at the synapse are translocated to the nucleus. A second gene discovered in the screen has opened for us the largely unstudied question of how synaptic boutons form. Rounded enlargements of presynaptic regions, i.e. synaptic boutons, have been described by neuroanatomists for over a century and they are a nearly universal feature of synapses, but their purpose is uncertain and the cytoskeletal arrangements and signaling pathways that cause them to form are not known. Our mutant screen identified two different genes that prevented bouton formation. One is in a protein called α2δ-3, previously known only as a subunit of Ca2+ channels. In null alleles of α2δ- 3, the motorneurons grow to the correct targets, stop there, put out thin neurites, and form active zones with clustered vesicles, but the neurites never round up to form proper boutons.
The other gene required for bouton formation was a member of the kinesin family of motors and a homolog of unc-104 and Kif1. Mutations in this gene, called immaculate connections, also give rise to neuromuscular contacts that never mature into boutons, but in this case the phenotype is more severe: the junctions also lack synaptic vesicles and have very low levels of active zone proteins. This kinesin is specific for cargos that are necessary for synaptogenesis and is not required for the outgrowth of the axon or navigation and target-recognition by the growth cone. We are trying to determine how this motor recognizes and binds the correct cargos and delivers them to the proper sites.
-
The Role of Ral and the Exocyst in Synaptic Plasticity:
Postsynaptic structures often have complex and characteristic shapes, although their functions are not always clear. Spines at excitatory CNS synapses are one of the most obvious and also one of great interest because their formation and retraction are linked to plastic changes in circuitry and behaviors. The growth of postsynaptic structures in response to activation will be a complex process involving changes in receptor traffic, cytoskeletal organization, and membrane expansion. We are presently studying the mechanism by which postsynaptic membranes expand and in particular the role of the small GTPase Ral in this process. Ral is a known regulator of the exocyst, a complex that mediates the targeting of vesicles to the correct regions of the cell surface for fusion. We have found that the complex folds of muscle membrane that surround boutons at the fly neuromuscular junction, a structure called the subsynaptic reticulum, is a plastic structure whose growth is regulated by the activity of the synapse. The influx of Ca 2+ upon synaptic activation activates Ral which in turn recruits the exocyst to the region of the SSR and mediates membrane addition. We are examining hippocampal neurons as well to see if a similar function for Ral may be important in membrane addition for spine growth. -
The Transport of Mitochondria in Axons and Dendrites and their Involvement in Neurodegenerative Disease:
Perhaps the most astonishing feature of neurons is the complexity of their shapes and in particular of their axons and dendritic trees. These processes may be a meter long in some human neurons, such as sensory and motor neurons or cortico- spinal tracts. Neurons developed these extraordinary extensions so that they could transmit electrical signals very rapidly over long distances. But this evolutionary adaptation came at a price: the need to transport supplies for equally long distances in order to keep these neurites supplied with proteins and organelles. This is particularly true for the cells mitochondria which need to supply energy and Ca2+ buffering to every part of the neuron. Indeed the task of distributing mitochondria within a neuron is very complex because they need to be present in every region of the cell in sufficient numbers to balance the energy demand in that region. Nor is this a one-time event in the cell; mitochondria need to be turned over and replaced just like every other cellular component. Indeed, damaged mitochondria may produce an excess of reactive oxygen species and need to be cleared away rapidly before inflicting further damage on the cell. In every cell, mitochondria move and fuse with one another and divide. For neurons, this is a matter of life and death.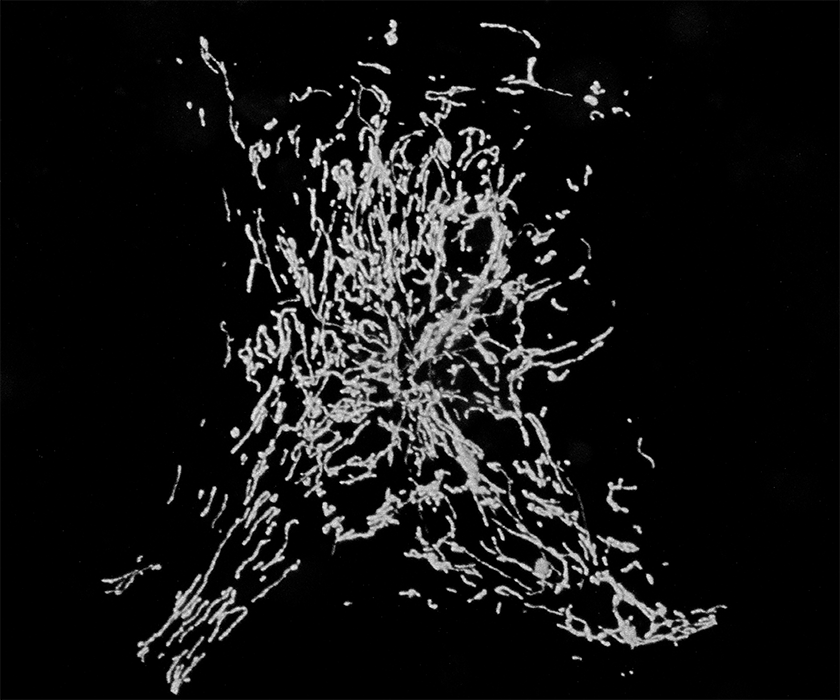
Our studies of a mutant fly we named milton led us to uncover a motor/adaptor complex needed for the transport of mitochondria in neurons and probably in most animal cells. The milton protein (which is also called TRAK1/2) binds to a regulatory protein on the mitochondrial surface called Miro (also called RhoT1/2). Milton also binds the motor proteins Kinesin Heavy Chain (Kif5) and thereby couples to the mitochondrial surface the motor that moves mitochondria to the plus-ends of microtubules, including moving them down axons.
With the elucidation of this motor/adaptor complex came the opportunity to study how the movement was regulated in order to match the needs of the cell. This includes studying how the motor for retrograde movement, dynein, is incorporated into this complex and how the complex is regulated by changes in cytosolic Ca2+ and by the enzyme O-GlcNAc Transferase (OGT). Ca2+ causes the mitochondria to halt their movement and this is probably a means of concentrating mitochondria in regions of high Ca2+ influx so that they can take up the excess Ca2+ and also provide ATP to help pump the ions back out. Failure in this mechanism makes neurons more susceptible to the type of excitotoxic insult experienced during ischemia. OGT is an enzyme that can transfer GlcNAc sugar residues to mitochondria and also halts their movement. OGT may allow mitochondria to sense the local nutrient supply and alter their distribution accordingly. We are currently investigating whether the overactivation of this pathway might contribute to the development of diabetic neuropathy.
One of the most intriguing regulations is the interaction of milton and Miro with two proteins that are mutated in hereditary forms of Parkinson's Disease: PINK1 and Parkin. These proteins form a pathway that is activated when mitochondria are damaged and lose their proper membrane potential. When the pathway is activated, the proteins bind to Miro and milton. PINK1 phosphorylates Miro which causes it to become degraded by the proteosome in a manner dependent on ubiquitination by Parkin. With the loss of Miro, mitochondria lose their motor proteins and stop. This arrest of motility is likely to prevent the damaged mitochondria from fusing with healthy mitochondria and, thus quarantined, allows them to be cleared away efficiently in a process called mitophagy. Having determined the mechanism by which these proteins arrest mitochondrial motility, we are presently characterizing the process by which they are subsequently cleared in axons.
Some of this work was described in a video we made to accompany an article in Cell. A link to that video can be found under the "Links" tab.
